
Au cours des cinq dernières années environ, les autorités sanitaires ont défini le cadre de la collecte de données structurées, plutôt que de simples documents, pour identifier et décrire les médicaments. La plus importante de ces normes est la suite de normes IDMP (Identification of Medicinal Products) développée par l’ISO. Ces normes sont mises en œuvre par l’EMA et contrôlées par la FDA.
Cette terminologie et ce format reconnus au niveau international facilitent l’interopérabilité des données, améliorent les activités de pharmacovigilance, évitent la duplication des activités de codage et permettent un échange d’informations plus facile avec et entre les autorités réglementaires. Alors pourquoi ne pas construire votre système de gestion de l’information réglementaire autour de l’IDMP ?
Vous voulez en savoir plus ? Téléchargez notre nouveau livre blanc, Considérations sur l’IDMP en 2022 : le rapport de données structurées interopérables (en anglais).
Le travail des entreprises pharmaceutiques n’est pas structurés en quatre domaines ISO faciles d’accès, comme le modèle utilisé dans l’IDMP. Bien que l’organisation ISO fournisse une référence utile, cette structure de données est conçue pour faciliter la collecte et le traitement de données structurées par une autorité réglementaire. Par conséquent, elle ne facilite pas les processus commerciaux des entreprises des sciences de la vie. Elle n’est pas structurée pour faciliter la saisie et l’extraction des données, ni pour l’examen et l’approbation des données.
Par exemple, l’IDMP n’a actuellement qu’une définition partielle en tant qu’identifiant de produit pharmaceutique, qui n’est pas encore entièrement décrite dans le guide de mise en œuvre (IG) de l’UE. Cependant, la réutilisation d’un produit et l’établissement de rapports sur celui-ci sont d’une importance capitale pour un promoteur pharmaceutique ou un MAH. Par conséquent, il n’est pas pratique de concevoir un système RIM autour de la définition de produit de l’IDMP.
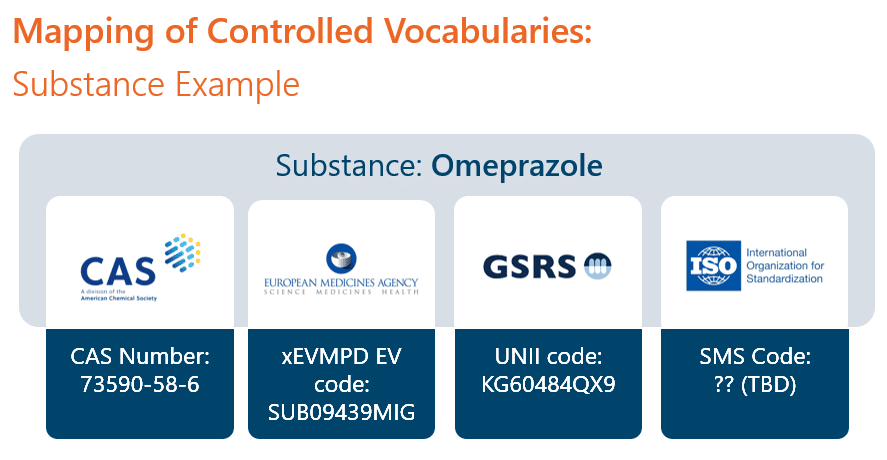
Un autre exemple concerne l’utilisation de vocabulaires contrôlés. Dans un système bien structuré, les termes contrôlés par l’entreprise doivent être mis en correspondance avec les termes et codes de déclaration des données structurées. Par exemple, une substance spécifique sera associée à un numéro CAS unique, qui sera ensuite utilisé dans les rapports CMC. La même substance sera également associée à un code xEVMPD EV qui sera soumis à l’EMA dans la composition. Si le même produit s’étend aux États-Unis, la substance doit correspondre au code UNII sous-jacent. L’IDMP fera correspondre la même substance à un code SMS. Dans un RIMS bien conçu, l’utilisateur saisit ou recherche le terme de l’entreprise pour la substance, et d’autres données correspondantes sont automatiquement disponibles et utilisées si nécessaire.
Heureusement, les données peuvent être modélisées dans un système RIM pharmaceutique pour combler ces lacunes. Les RIMS doivent être indépendants mais conformes aux contraintes des directives réglementaires en termes d’objets, de définition des classes et de relations entre les données (cardinalité). Ceci veut dire que si votre RIMS est bien structuré, la création du message de données structurées pour le reporting est une simple traduction de vos termes acceptés en interne. Lorsque les directives changent, votre système RIM peut s’y adapter en modifiant simplement la cartographie ou en ajoutant des éléments de données.
Si l’on suit ce raisonnement, les directives réglementaires évolueront au fil du temps, mais elles ne doivent pas être autorisées à dicter la structure des données de base du RIM. Après tout, les directives relatives au format structuré et aux rapports témoignent du point de vue de l’autorité réglementaire, et non du promoteur.
Vous voulez en savoir plus ? Téléchargez notre nouveau livre blanc, Considérations sur l’IDMP en 2022 : le rapport de données structurées interopérables (en anglais).